 TOC |
HEME
TOC |
HEME
Mayo Clin Proc. 2005;80:75-83 © 2005 Mayo Foundation for Medical Education
and Research
http://www.mayoclinicproceedings.com/inside.asp?AID=821&UID=3926
REVIEW
Blood Eosinophilia: A New Paradigm in Disease Classification, Diagnosis,
and Treatment
From the Department of Internal Medicine and Division
of Hematology, Mayo Clinic College of Medicine, Rochester, Minn.
AYALEW TEFFERI, MD
Address reprint requests and correspondence to Ayalew Tefferi, MD, Division
of Hematology, Mayo Clinic College of Medicine, 200 First St SW, Rochester,
MN 55905 (e-mail:
tefferi.ayalew@mayo.edu).
Abstract
Acquired blood eosinophilia is considered either a primary or a secondary
phenomenon. Causes of secondary (ie, reactive) eosinophilia include
tissue-invasive parasitosis, allergic or inflammatory conditions, and
malignancies in which eosinophils are not considered part of the neoplastic
process. Primary eosinophilia is classified operationally into 2 categories:
clonal and idiopathic. Clonal eosinophilia stipulates the presence of either
cytogenetic evidence or bone marrow histological evidence of an otherwise
classified hematologic malignancy such as acute leukemia or a chronic myeloid
disorder. Idiopathic eosinophilia is a diagnosis of exclusion (ie, not secondary
or clonal). Hypereosinophilic syndrome is a subcategory of idiopathic
eosinophilia; diagnosis requires documentation of both sustained eosinophilia
(absolute eosinophil count =1500 cells/µL for at least 6 months) and
target organ damage (eg, involvement of the heart, lung, skin, or nerve tissue).
Genetic mutations involving the platelet-derived growth factor receptor genes
(PDGFR-a and PDGFR-ß) have been pathogenetically linked to clonal
eosinophilia, and their presence predicts treatment response to imatinib.
Accordingly, cytogenetic and/or molecular investigations for the presence
of an imatinibsensitive molecular target should accompany current evaluation
for primary eosinophilia. In the absence of such a drug target, specific
treatment is dictated by the underlying hematologic malignancy in cases of
clonal eosinophilia; however, the initial treatment of choice for symptomatic
patients with hypereosinophilic syndrome is prednisone and/or interferon
alfa.
Mayo Clin Proc. 2005;80(1):75-83
| AEC = absolute eosinophil count; CEL = chronic eosinophilic
leukemia; CML = chronic myeloid leukemia; CMML = chronic myelomonocytic
leukemia; FGFR1 = fibroblast growth factor receptor 1; FISH =
fluorescence in situ hybridization; HES = hypereosinophilic
syndrome; IL = interleukin; MPD = myeloproliferative
disorder; PDGFR = platelet-derived growth factor receptor; SM =
systemic mastocytosis; SM-eos = SM associated with prominent blood
eosinophilia |
Eosinophils are derived from hematopoietic stem cells that are committed
initially to the myeloid and subsequently to the basophil-eosinophil granulocyte
lineage.1 As depicted in
Figure 1, the mature eosinophil, when examined
by standard blood-staining techniques, displays a bilobed nucleus and an
abundant cytoplasm filled with reddish-orange granules. The material in these
granules includes cationic proteins (major basic protein, eosinophilic cationic
protein, eosinophil-derived neurotoxin, eosinophil peroxidase), cytokines
(interleukins [ILs], tumor necrosis factor), and lipid mediators (leukotriene
C4).2 Interleukin 5 is considered
the major eosinophil growth and survival factor, whereas chemokines (eotaxin,
platelet-activating factor, RANTES [regulated on activation, normal T expressed
and secreted]) and endothelial adhesion molecules (integrins, vascular cell
adhesion molecules) contribute to eosinophil
trafficking.3-5
Major basic protein, eosinophil cationic protein, and eosinophil-derived
neurotoxin are the primary mediators of eosinophil-associated toxicity to
microbes (parasites, protozoa, bacteria, viruses) and human tissue (myocarditis,
pneumonitis, dermatitis, neuropathy,
vasculitis).6-16 The
lung and gastrointestinal systems constitute the main residence for eosinophils.
Blood eosinophilia (absolute eosinophil count [AEC] =600 cells/µL) is
the usual initial clue for the presence of an eosinophilic disorder. The
degree of blood eosinophilia, in the absence of active treatment, can be
categorized into mild (AEC 600-1500 cells/µL), moderate (AEC 1500-5000
cells/µL), or severe (AEC >5000
cells/µL).17 Target organ damage is
unusual with mild eosinophilia, but its occurrence in association with moderate
to severe eosinophilia does not appear to depend on the specific cause of
eosinophilia (ie, in addition to the well-established association of target
organ damage with primary eosinophilia, it also has been reported to occur
in both familial and secondary
eosinophilia).18,19
GENERAL CLASSIFICATION OF EOSINOPHILIC
DISORDERS
Blood eosinophilia can be classified as either familial or acquired
(Table 1). Acquired eosinophilia is classified
further into a primary or a secondary process, depending on whether eosinophils
are considered integral to the underlying disease. Infectious causes and
noninfectious causes of secondary eosinophilia (Table
1) are prevalent in underdeveloped and developed countries, respectively.
Primary eosinophilia occurs primarily in males and is considered clonal in
the presence of either cytogenetic evidence or bone marrow histological evidence
of an otherwise defined neoplastic hematologic disorder. Otherwise, a working
diagnosis of idiopathic eosinophilia is made, and in the presence of both
sustained eosinophilia (AEC =1500 cells/µL for at least 6 months) and
target organ damage (eg, skin, heart, lung, nerve tissue), the process is
subclassified as hypereosinophilic syndrome
(HES).20
 eosinophilia_cell.jpg
eosinophilia_cell.jpg
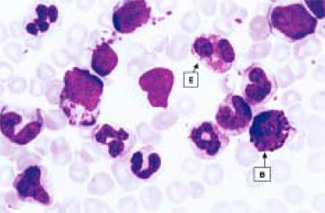
FIGURE 1. Peripheral blood smear shows an eosinophil (E), basophil (B), and
other blood cells (image courtesy of Chin-Yang Li, MD, Department of Laboratory
Medicine and Pathology, Division of Hematopathology, Mayo Clinic, Rochester,
Minn). |
TABLE 1. Classification and Causes of Blood
Eosinophilia*
-
Familial
-
Acquired
-
Secondary
-
Infectious causes
-
Tissue-invasive parasitosis (most common)
-
Bacterial or viral infections (rare)
-
Noninfectious causes
-
Drugs (sulfa, carbamazepine, etc)
-
Toxins (associated with eosinophilia-myalgia syndrome, toxic oil syndrome,
etc)
-
Allergy (asthma, atopic dermatitis, etc)
-
Idiopathic/autoimmune inflammatory conditions
-
Vasculitis (Churg-Strauss, Wegener)
-
Kimura disease
-
Eosinophilic fasciitis
-
Systemic sclerosis or scleroderma
-
Polyarteritis and other CTDs
-
Sarcoidosis
-
Inflammatory bowel disease
-
Malignancy (metastatic cancer, Hodgkin lymphoma)
-
Endocrinopathies (Addison disease, etc)
-
Primary
-
Clonal
-
Acute leukemia
-
Acute myeloid leukemia
-
Acute lymphocytic leukemia
-
Chronic myeloid disorder
-
Molecularly defined chronic myeloid disorder
-
Bcr/Abl+ chronic myeloid leukemia
-
PDGFRA-rearranged eosinophilic disorder
-
PDGFRB-rearranged eosinophilic disorder
-
Kit-mutated systemic mastocytosis
-
8p11 Syndrome
-
Clinicopathologically defined chronic myeloid disorder
-
Myelodysplastic syndrome
-
Myeloproliferative disorder
-
Classic myeloproliferative disorder (polycythemia vera, etc)
-
Atypical myeloproliferative disorder
-
Chronic eosinophilic leukemia
-
Systemic mastocytosis
-
Chronic myelomonocytic leukemia
-
Unclassified myeloproliferative disorder
-
Idiopathic
-
HES
-
Pre-HES
*CTD = connective tissue disease; HES = hypereosinophilic syndrome; PDGFR
= platelet-derived growth factor receptor.
PATHOGENESIS AND CLINICAL
MANIFESTATIONS
FAMILIAL EOSINOPHILIA
Familial eosinophilia is rare, and its genetic basis, at least in some cases,
may be similar to that of clonal
eosinophilia.18,21 In
such cases, the chromosomal region 5q31-33 has been identified as the “hot
zone,” although it may not involve mutations of some of the resident
genes responsible for the synthesis of known eosinophil growth factors including
IL-3, IL-5, and granulocyte-macrophage colony-stimulating
factor.22 Interestingly, one recently reported
familial case involved a translocation-associated mutation of the
platelet-derived growth factor receptor gene PDGFRB, which is known
to reside in the 5q31-33 chromosomal region and to be pathogenetically linked
to some cases of clonal
eosinophilia.21,23 In
general, familial eosinophilia is an extremely rare autosomal dominant disorder
that is characterized by a stable eosinophil count and, compared with HES,
a lesser degree of eosinophil activation as well as a more benign clinical
course.24
ACQUIRED EOSINOPHILIA
Secondary Eosinophilia.
The most common cause of secondary eosinophilia is tissue-invasive parasitosis
including schistosomiasis, visceral toxocariasis, strongyloidiasis, filariasis,
ancylostomiasis, fascioliasis, trichinellosis, and paragonimiasis
(Table
1).25-32 In general,
parasites that are isolated in either the intestinal lumen (Cestoda,
Ascaris) or an intact cyst (Echinococcus granulosus) do not
cause blood eosinophilia unless they are introduced systemically through
tissue invasion or cyst disruption.33 In
addition to helminths, some (Toxoplasma gondii, Dientamoeba fragilis,
Isospora belli) but not other (Giardia lamblia, Entamoeba
histolytica) protozoan infections may induce blood
eosinophilia.33-35 Rare
reports of eosinophilia-associated bacterial (borrelia) or viral (human
immunodeficiency virus) infections have been
published.36,37
Noninfectious causes of secondary eosinophilia include drugs (sulfa derivatives,
gold compounds, carbamazepine, myeloid growth factors, purine nucleoside
analogues),38,45 toxins
(associated with eosinophilia-myalgia syndrome, toxic oil syndrome, toxic
shock
syndrome),46,48 allergic
disorders (asthma, hay fever, atopic dermatitis, allergic bronchopulmonary
aspergillosis),49-51
idiopathic/autoimmune inflammatory conditions (granulomatous and/or systemic
vasculitis, Churg-Strauss syndrome, Wegener granulomatosis, Kimura disease,
angiolymphoid hyperplasia with eosinophilia, bullous pemphigoid and other
cutaneous disorders, eosinophilic fasciitis, systemic sclerosis or scleroderma,
polyarteritis, sarcoidosis, inflammatory bowel disease), malignancies in
which eosinophils are not considered part of the neoplastic clone (Hodgkin
and non-Hodgkin lymphoma, metastatic
cancer),52-57 and
endocrinopathies including Addison disease (Table
1).58 Secondary eosinophilia in inflammatory
and malignant conditions is mediated by tissue-derived or tumor-derived
eosinophilogenic
cytokines.56,57,59-61
Primary Eosinophilia.
Clonal
Eosinophilia.
A diagnosis of clonal eosinophilia requires the presence of either cytogenetic
evidence or bone marrow morphologic evidence for either acute leukemia or
a chronic myeloid disorder (Table 1). Hematologic
disorders that can be accompanied by clonal eosinophilia include acute myeloid
leukemia,62 acute lymphocytic
leukemia,63 chronic myeloid leukemia
(CML),64 myelodysplastic
syndrome,65 and both classic and atypical
cases of myeloproliferative disorders
(MPDs).66,67 The atypical
MPD category includes chronic eosinophilic leukemia
(CEL),68 systemic mastocytosis
(SM),69 and chronic myelomonocytic leukemia
(CMML).70 Recent developments in the molecular
characterization of pathogenesis, in a subset of patients with clonal
eosinophilia, have identified activating mutations of 3 receptor tyrosine
kinase genes: PDGFRA, PDGFRB, and fibroblast growth factor
receptor 1 (FGFR1).71
Within the context of hematologic malignancies, PDGFRA mutations have
been connected so far with a subset of patients with SM associated with prominent
blood eosinophilia
(SM-eos).72-76 In general,
SM-eos makes up approximately 20% of all cases of SM in
adults.77 Of these cases of SM-eos, approximately
half may be associated with a specific PDGFRA mutation caused by an
800 kilobase interstitial deletion involving chromosome 4q12, resulting in
juxtaposition of PDGFRA and FIP1L1 genes
(FIP1L1-PDGFRA).74,75
The fusion gene generates a constitutively activated PDGFRA tyrosine kinase
that transforms hematopoietic cells.75 Other
cases of SM-eos are associated with a C-kit
mutation.74 Systemic mastocytosis not associated
with eosinophilia does not display the FIP1L1-PDGFRA
mutation.75 Although rare, PDGFRA
also may be activated through chromosomal translocations as in
t(4;22)(q12;q11).76
Patients with FIP1L1-PDGFRA+ SM-eos display a
“myeloproliferative” clinical phenotype with organomegaly, cytopenia,
increased serum B12 levels, and bone marrow hypercellularity with
myelofibrosis.75,78,79
As a result, investigators use the terms myeloproliferative-variant
HES78 and
FIP1L1-PDGFRA+CEL79
to describe the same disease. However, most, if not all, such patients display
bone marrow mast cell infiltration with morphologically and immunophenotypically
abnormal mast
cells.72,73 Regardless,
the term FIP1L1-PDGFRA+eosinophilic disorder
could be used to avoid confusion.73 Patients
with FIP1L1-PDGFRA+ eosinophilic disorder also are
characterized by a high risk of eosinophilic heart disease and inferior survival
compared with those with
FIP1L1-PDGFRA– clonal
eosinophilia.75,79
The PDGFRB gene is located on chromosome 5q33, and its activation
through translocation to various partner chromosomes, including t(5;12)(q33;p13),
t(5;10)(q33;q21), t(5;7)(q33;q11.2), t(5;14)(q33;q13), t(5;17)(q33;p11),
and t(1;5)(q23;q33), produces a distinct MPD that usually is associated with
prominent eosinophilia and sometimes with
monocytosis.23,66,80
Pathologic diagnosis in previously described cases of
PDGFRB+ clonal eosinophilia have included CEL, atypical
CML or MPD, and
CMML.81-83
The gene for FGFR1 is located on chromosome 8p11, and its activation through
translocation to different chromosome partners, including t(8;13)(p11;q12),
t(8;9)(p11;q33), t(6;8)(q27;p11), and t(8;22)(p11;q22), produces a
myeloproliferative syndrome with eosinophilia, lymphadenopathy, and a high
incidence of T-cell non-Hodgkin lymphoma with progression to acute myeloid
leukemia.84 Other karyotypic variants involving
FGFR1, including t(8;17)(p11;q25), t(8;11)(p11;p15), t(8;12)(p11;q15),
and ins(12;8)(p11;p11; p21), have been associated with SM, acute leukemia,
or MPD associated with both T-cell lymphoma and marrow
eosinophilia.85
Idiopathic
Eosinophilia.
When both clinical and laboratory evaluations do not clearly identify either
a secondary or a clonal cause, a working diagnosis of idiopathic eosinophilia
is reasonable. For now, HES is considered a subcategory of idiopathic
eosinophilia that requires documentation of both target organ damage and
an AEC of 1500 cells/µL or greater for at least 6
months.20,86 Several
review articles have described the clinical manifestations and natural history
of
HES.20,86,88
These and other case reports have shown that some cases evolve into either
acute leukemia or an aggressive disease phenotype that is indistinguishable
from an
MPD.89-91 This information
has long suggested that HES may represent a clonal hematologic disorder.
This contention was supported by clonality studies that used X-linked DNA
analysis.92 In contrast, the presence of
clonal TH2 lymphocytes in a subset of patients with “HES”
suggests a pathogenetic heterogeneity that may include an IL-5–mediated
secondary eosinophilia in the
mix.93,94 In a study
of 60 patients with HES, 16 (27%) had T cells with an aberrant immunophenotype,
and T-cell clonality was suggested in half of these 16
patients.95
As in clonal eosinophilia, more than 90% of patients with HES are males.
The reason for male predominance in HES and in clonal eosinophilia is unknown.
Clinical manifestations can be either nonspecific (cough, nocturnal sweating,
fatigue, anorexia, weight loss, gastrointestinal
symptoms)96 or directly related to an affected
target organ including the skin (pruritus, erythematous papules, nodules,
urticaria, angioedema, mucosal ulcers, eosinophilic cellulitis or Wells
syndrome),97-99 heart
(mural thrombus, endocardial fibrosis, cardiomyopathy, elevated serum troponin
level),86,100,101
nervous system (sensory and motor neuropathy, mononeuritis multiplex, isolated
vasculitis of central nervous system, eosinophilic meningitis, transverse
myelitis),102-105
lung (pulmonary infiltrates, lung nodules, acute respiratory distress
syndrome),106-108
or gastrointestinal system (gastroenteritis, inflammatory bowel disease,
sclerosing
cholangitis).109-112
Thromboembolic complications, including Budd-Chiari syndrome, are not infrequent
in
HES,96,113,114
and in isolated reports, HES has been associated with retinal vein occlusions,
polymyositis, arthritis, and renal
disease.115-118
DIAGNOSIS
A thorough patient history is the most important part of the evaluation for
blood eosinophilia, and it should guide the extent and type of laboratory
tests performed. The initial focus should be on international travel history
and a review of current and recent medications. In the absence of possible
drug association, a stool test should be performed in all cases to look for
ova and larvae of intestinal worms (ascaris, schistosoma, ancylostoma, cestodes,
fasciola, strongyloides). Next, depending on the travel history, the following
tests and procedures should be performed: urine sediment tests (schistosoma),
blood concentration tests (filaria), serology (strongyloides, filaria,
trichinosis, visceral larva migrans, schistosomiasis), sputum examination
(paragonimiasis, visceral larva migrans), chest radiography (paragonimiasis,
ascariasis), computed tomography (echinococcus, cysticercosis), small bowel
biopsy (isosporiasis, strongyloidosis), or muscle biopsy (trichinosis). However,
work-up beyond stool examination is recommended only if history dictates.
Similarly, a good history and basic laboratory tests should be adequate to
address the possibility of a noninfectious cause of secondary eosinophilia,
and only in the presence of historical clues should more invasive and costly
interventions be pursued.
All patients with suspected primary eosinophilia should undergo bone marrow
examination with cytogenetics. Also, my colleagues and I encourage use of
bone marrow immunohistochemical stains for tryptase and mast cell
immunophenotyping (neoplastic but not normal or reactive mast cells express
CD25) to test for SM. The following PDGFRB rearrangements currently
are detected by conventional cytogenetics: t(5;12)(q33;p13), t(5;10)(q33;q21),
t(5;7)(q33;q11.2), t(5;14)(q33;q13), t(5;17)(q33;p11), and t(1;5)(q23;q33).
In contrast, standard cytogenetic methods do not detect the usual
PDGFRA rearrangement that is a result of an interstitial 4q12 deletion.
This must be sought with either a fluorescence in situ hybridization
(FISH)–based or a reverse transcriptase polymerase chain
reaction–based laboratory
technique.73 At present, we perform 3 laboratory
tests (bone marrow biopsy, bone marrow cytogenetics, peripheral blood FISH
for FIP1L1-PDGFRA) in all patients with primary eosinophilia because
of the important therapeutic implications (Figure
2).
Finally, in addition to looking for the cause of eosinophilia, laboratory
tests to assess possible eosinophilic tissue damage may be required and include
echocardiography, chest radiography, pulmonary function tests, and, in the
presence of symptoms, tissue biopsy.
TREATMENT OF PRIMARY
EOSINOPHILIA
CLONAL EOSINOPHILIA
In general, the underlying hematologic malignancy dictates treatment in clonal
eosinophilia. In acute leukemia, for example, standard induction chemotherapy
is used.119 In CML, imatinib is the drug
of choice.120 Imatinib works in CML by
inhibiting the leukemogenic bcr-abl gene product, which is a
constitutively activated Abl tyrosine
kinase.121-123 Although
relatively selective in its action, imatinib inhibits other receptor tyrosine
kinases including PDGFR and
Kit.124,125 The mechanism
of action involves a competitive occupation of an adenosine
triphosphate–binding site of the catalytic domain of the kinase by a
conformation-dependent mechanism.126
The in vitro demonstration of imatinib-induced inhibition of both PDGFR-
and Kit-associated signal transduction formed the rationale for using the
drug in clonal eosinophilia associated with SM (SM-eos). This is because
(1) Kit ligand and Kit-associated signaling are key for the growth and
development of human mast cells127 and (2)
approximately 50% of patients with SM-eos may carry a constitutively activated
PDGFRA mutation, as discussed
previously.72,75 In
vitro, imatinib effectively inhibits normal mast cell growth and
development128 as well as the growth of
human mast cell lines and primary cells that carry certain (Val560Gly) but
not other (Asp816Val) c-kit
mutations.129,130
Consistent with these in vitro observations, imatinib has been shown effective
in a subset of patients with SM-eos who carry the FIP1L1-PDGFRA mutation
but not the c-kit Asp816Val
mutation.131 Patients with SM-eos and the
FIP1L1-PDGFRA mutation experience both molecular and mast cell
immunophenotypic remissions with relatively low doses of imatinib (100
mg/d).74,132 Other
investigators refer to FIP1L1-PDGFRA+ SM-eos as
either myeloproliferative-variant HES,
FIP1L1-PDGFRA+ CEL, or
FIP1L1-PDGFRA+ HES and have reported equally impressive
imatinib treatment
results.75,78,79,133-135 Imatinib therapy produces
complete clinical, pathologic, and molecular remissions in clonal eosinophilia
connected with PDGFRB mutations that are usually associated with a
chronic myeloid disorder that phenotypically resembles CEL, CMML, or atypical
MPD.66,82,136

FIGURE 2. A diagnostic and treatment algorithm for blood eosinophilia. FISH
= fluorescence in situ hybridization; RT-PCR = reverse transcriptase polymerase
chain reaction. |
IDIOPATHIC EOSINOPHILIA INCLUDING HES
Most patients with symptomatic HES respond to prednisone alone or in combination
with
hydroxyurea.137,138
Patients with HES refractory to corticosteroids have been reported to respond
to cyclosporine,139
vincristine,140,141
interferon
alfa,142-144
cladribine,141,145
or etoposide.146 My preference during
symptomatic disease is to use interferon alfa early either by itself or combined
with low doses of prednisone. For patients in whom first-line treatment with
corticosteroids, hydroxyurea, and interferon alfa fails, the choice of
second-line treatment is currently arbitrary, and any of the aforementioned
agents can be used before an aggressive approach such as allogeneic hematopoietic
stem cell transplantation is considered seriously. Because of the documentation
of imatinib-induced partial remissions in approximately 50% of patients with
true HES (ie, FIP1L1-PDGFRA–), it is reasonable
to try the specific drug as a second-line
therapy.75 However, it should be noted that
patients with true HES are unlikely to respond to low-dose imatinib (100
mg/d), and a higher dose of the drug (400 mg/d) should be used in this instance
(Figure 2). In patients with HES refractory to
drugs, both myeloablative and non-myeloablative allogeneic hematopoietic
stem cell transplantation have been used successfully and can be
considered.147-150
Currently, the role of novel drug treatment approaches to HES, including
the use of
imatinib75,78,79
and monoclonal antibodies to either
IL-5151-153 or CD52
(alemtuzumab),154 is being defined.
CURRENT ALGORITHM FOR THE DIAGNOSIS AND TREATMENT OF EOSINOPHILIC
DISORDERS
Figure 2 outlines a proposed algorithm for the
diagnosis and treatment of eosinophilic disorders. To begin treatment of
a patient with blood eosinophilia, the possibility of secondary eosinophilia
must be excluded. Once this is accomplished, I recommend obtaining a set
of blood and bone marrow studies. The blood studies should include serum
tryptase (an increased level suggests SM), T-cell receptor gene rearrangement
analysis (positive test results suggest an underlying clonal T-cell disorder),
and serum IL-5 (an elevated level requires careful evaluation of bone marrow
studies and T-cell gene rearrangement studies for the presence of a clonal
T-cell disease). Bone marrow examination should include cytogenetic studies,
tryptase immunostains, and FISH or reverse transcriptase polymerase chain
reaction to screen for FIP1L1-PDGFRA. The last-mentioned test also
can be performed on peripheral blood.
If this work-up reveals an imatinib-sensitive molecular target (PDGFRA
or PDGFRB mutations), then treatment with low-dose imatinib (100 mg/d)
is recommended, and a complete and durable remission can be expected. We
currently recommend an initial imatinib dosage of 100 mg/d for
FIP1L1-PDGFRA+ clonal eosinophilia because this
specific dosage, as discussed previously, has been associated with molecular
disease remission.73 It is reasonable to
consider treatment even in the absence of symptoms for
FIP1L1-PDGFRA+ clonal eosinophilia in hopes of
preventing serious cardiovascular complications that may or may not be amenable
to imatinib therapy once they
occur.78,155
In the absence of an imatinib-sensitive molecular target, the physician first
must decide whether treatment is indicated and if so, choose a first-line
treatment that is suitable for the underlying clonal process or, in cases
of HES, treat with interferon alfa and/or prednisone. Imatinib is unlikely
to benefit patients with clonal eosinophilia who do not have a known
drug-sensitive mutation. Similarly, although some patients with true HES
experience partial remission with imatinib therapy at 400
mg/d,72 the long-term benefit of single-agent
imatinib therapy in FIP1L1-PDGFRA– HES is unknown.
Finally, we and others have described life-threatening cardiogenic shock
associated with imatinib therapy for
HES.156,157 Fortunately,
this potentially fatal complication responds well to systemic corticosteroid
therapy and may be predicted by elevated serum troponin levels before
treatment.157 For patients with either baseline
elevation of serum troponin level or echocardiographic evidence for eosinophilic
heart disease, we recommend concomitant prednisone therapy (1 mg/kg) during
the first week of treatment with imatinib. Prednisone can be tapered during
the second week of therapy.
CONCLUSION
The clinical observation158 that some patients
with “HES” responded to treatment with imatinib, which is a small
molecule inhibitor of certain receptor tyrosine
kinases,73 led to the recent discovery of
the association between blood eosinophilia and a cytogenetically occult genetic
mutation (FIP1L1-PDGFRA).75 Such molecular
characterizations may ultimately result in HES becoming a nonentity in the
spectrum of primary eosinophilic disorders. More importantly, the identification
of disease-causing mutations is an essential step toward the development
and application of rational drug therapy. At present, approximately 10% to
20% of patients with primary eosinophilia are known to harbor a molecular
target that predicts an excellent response to imatinib
therapy.72,73 However,
a subset of patients with HES without known imatinib targets also has been
reported to respond to the
drug.73,75 These
observations raise the exciting prospect of discovering new drug targets
in eosinophilic disorders as well as future therapeutic successes with the
use of second-generation kinase inhibitors that are more potent than imatinib
and have a broader spectrum of
activity.159,160
REFERENCES
-
Denburg JA, Telizyn S, Messner H, et
al. Heterogeneity of human peripheral blood eosinophil-type colonies:
evidence for a common basophileosinophil
progenitor. Blood. 1985;66:312-318.
-
Rothenberg ME. Eosinophilia. N
Engl J Med. 1998;338:1592-1600.
-
Yamaguchi Y, Hayashi Y, Sugama Y, et
al. Highly purified murine interleukin 5 (IL-5) stimulates eosinophil
function and prolongs in vitro survival: IL-5 as an eosinophil chemotactic
factor. J Exp Med. 1988;167:1737-1742.
-
Elsner J, Kapp A.
The chemokine network in eosinophil activation. Allergy Asthma
Proc. 2001;22:139-148.
-
Dobrina A, Menegazzi R, Carlos TM, et
al. Mechanisms of eosinophil adherence to cultured vascular endothelial
cells: eosinophils bind to the cytokine-induced ligand vascular cell adhesion
molecule-1 via the very late activation antigen-4 integrin receptor. J
Clin Invest. 1991;88:20-26.
-
Gleich GJ, Adolphson CR, Leiferman KM.
The biology of the eosinophilic leukocyte. Annu Rev
Med. 1993;44:85-101.
-
Hamann KJ, Gleich GJ, Checkel JL, Loegering DA, McCall JW, Barker RL.
In vitro killing of microfilariae of Brugia pahangi and Brugia
malayi by eosinophil granule proteins. J
Immunol. 1990;144:3166-3173.
-
Lehrer RI, Szklarek D, Barton A, Ganz T, Hamann KJ, Gleich GJ.
Antibacterial properties of eosinophil major basic protein and eosinophil
cationic protein. J Immunol. 1989;142:4428-4434.
-
Torpier G, Colombel JF, Mathieu-Chandelier C, et
al. Eosinophilic gastroenteritis: ultrastructural evidence for a selective
release of eosinophil major basic protein. Clin Exp
Immunol. 1988;74:404-408.
-
Tai PC, Ackerman SJ, Spry CJ, Dunnette S, Olsen EG, Gleich GJ.
Deposits of eosinophil granule proteins in cardiac tissues of patients
with eosinophilic endomyocardial
disease. Lancet. 1987;1:643-647.
-
Slifman NR, Loegering DA, McKean DJ, Gleich GJ.
Ribonuclease activity associated with human eosinophil-derived neurotoxin
and eosinophil cationic protein. J
Immunol. 1986;137:2913-2917.
-
Young JD, Peterson CG, Venge P, Cohn ZA.
Mechanism of membrane damage mediated by human eosinophil cationic
protein. Nature. 1986;321:613-616.
-
Sorrentino S, Glitz DG, Hamann KJ, Loegering DA, Checkel JL, Gleich GJ.
Eosinophil-derived neurotoxin and human liver ribonuclease: identity
of structure and linkage of neurotoxicity to nuclease activity. J
Biol Chem. 1992;267:14859-14865.
-
Sunohara N, Furukawa S, Nishio T, Mukoyama M, Satoyoshi E.
Neurotoxicity of human eosinophils towards peripheral nerves. J
Neurol Sci. 1989;92:1-7.
-
Chen KR, Su WP, Pittelkow MR, Conn DL, George T, Leiferman KM.
Eosinophilic vasculitis in connective tissue disease. J Am
Acad Dermatol. 1996;35(2, pt 1):173-182.
-
Shah AM, Brutsaert DL, Meulemans AL, Andries LJ, Capron M.
Eosinophils from hypereosinophilic patients damage endocardium of isolated
feline heart muscle
preparations. Circulation. 1990;81:1081-1088.
-
Brito-Babapulle F. The eosinophilias,
including the idiopathic hypereosinophilic syndrome. Br J
Haematol. 2003;121:203-223.
-
Lin AY, Nutman TB, Kaslow D, et
al. Familial eosinophilia: clinical and laboratory results on a
U.S.kindred. Am J Med Genet. 1998;76:229-237.
-
Yakulis R, Bedetti CD.
Loffler’s endocarditis: occurrence with malignant lymphoma with
a high content of epithelioid histiocytes (‘Lennert’s
lymphoma’). Arch Pathol Lab Med. 1983;107:531-534.
-
Chusid MJ, Dale DC, West BC, Wolff SM.
The hypereosinophilic syndrome: analysis of fourteen cases with review
of the literature. Medicine (Baltimore). 1975;54:1-27.
-
Bakhshi S, Hamre M, Mohamed AN, Feldman G, Ravindranath Y.
t(5;9)(q11;q34): a novel familial translocation involving Abelson oncogene
and association with hypereosinophilia. J Pediatr Hematol
Oncol. 2003;25:82-84.
-
Rioux JD, Stone VA, Daly MJ, et
al. Familial eosinophilia maps to the cytokine gene cluster on human
chromosomal region 5q31-q33. Am J Hum
Genet. 1998;63:1086-1094.
-
Baxter EJ, Kulkarni S, Vizmanos J-L, et
al. Novel translocations that disrupt the platelet-derived growth factor
receptor ß (PDGFRB) gene in BCR–ABL-negative chronic
myeloproliferative disorders. Br J
Haematol. 2003;120:251-256.
-
Klion AD, Law MA, Riemenschneider W, et
al. Familial eosinophilia: a benign
disorder? Blood. 2004;103:4050-4055.
-
Evengard B. Diagnostic and clinical
aspects of schistosomiasis in 182 patients treated at a Swedish ward for
tropical diseases during a 10-year period. Scand J Infect
Dis. 1990;22:585-594.
-
Zinkham WH. Visceral larva migrans:
a review and reassessment indicating two forms of clinical expression: visceral
and ocular. Am J Dis Child. 1978;132:627-633.
-
Milder JE, Walzer PD, Kilgore G, Rutherford I, Klein M.
Clinical features of Strongyloides stercoralis infection in
an endemic area of the United
States. Gastroenterology. 1981;80:1481-1488.
-
Hussain R, Hamilton RG, Kumaraswami V, Adkinson NFJr, Ottesen EA.
IgE responses in human filariasis, I: quantitation of filaria-specific
IgE. J Immunol. 1981;127:1623-1629.
-
Maxwell C, Hussain R, Nutman TB, et
al. The clinical and immunologic responses of normal human volunteers
to low dose hookworm (Necator americanus) infection. Am J
Trop Med Hyg. 1987;37:126-134.
-
Arjona R, Riancho JA, Aguado JM, Salesa R, Gonzalez-Macias J.
Fascioliasis in developed countries: a review of classic and aberrant
forms of the disease. Medicine
(Baltimore). 1995;74:13-23.
-
Pozio E, Varese P, Morales MA, Croppo GP, Pelliccia D, Bruschi F.
Comparison of human trichinellosis caused by Trichinella spiralis
and by Trichinella britovi. Am J Trop Med
Hyg. 1993;48:568-575.
-
Uchiyama F, Morimoto Y, Nawa Y.
Re-emergence of paragonimiasis in Kyushu, Japan. Southeast
Asian J Trop Med Public Health. 1999;30:686-691.
-
Windsor JJ, Johnson EH.
Dientamoeba fragilis: the unflagellated human
flagellate. Br J Biomed Sci. 1999;56:293-306.
-
Grant SC, Klein C.
Toxoplasma gondii encephalitis in an immunocompetent adult:
a case report. S Afr Med J. 1987;71:585-587.
-
Junod C. Isospora belli coccidiosis
in immunocompetent subjects (a study of 40 cases seen in Paris) [in
French]. Bull Soc Pathol Exot
Filiales. 1988;81:317-325.
-
Granter SR, Barnhill RL, Duray PH.
Borrelial fasciitis: diffuse fasciitis and peripheral eosinophilia
associated with Borrelia infection. Am J
Dermatopathol. 1996;18:465-473.
-
Tietz A, Sponagel L, Erb P, Bucher H, Battegay M, Zimmerli W.
Eosinophilia in patients infected with the human immunodeficiency
virus. Eur J Clin Microbiol Infect
Dis. 1997;16:675-677.
-
Spry CJ. Eosinophilia and allergic
reactions to drugs. Clin Haematol. 1980;9:521-534.
-
Tas S, Simonart T.
Management of drug rash with eosinophilia and systemic symptoms (DRESS
syndrome): an update. Dermatology. 2003;206:353-356.
-
Ichiche M, Kiesch N, De
Bels D. DRESS syndrome associated with HHV-6
reactivation. Eur J Intern Med. 2003;14:498-500.
-
De
Marco P, Melchiori G.
Carbamazepine and eosinophilia [letter]. Ann
Neurol. 1986;20:274.
-
Gonzales-Chambers R, Rosenfeld C, Winkelstein A, Dameshek L.
Eosinophilia resulting from administration of recombinant
granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in a patient
with T-gamma lymphoproliferative disease. Am J
Hematol. 1991;36:157-159.
-
Seebach J, Speich R, Fehr J, Tuchschmid P, Russi E.
GM-CSF-induced acute eosinophilic pneumonia. Br J
Haematol. 1995;90:963-965.
-
Trojan A, Meier R, Licht A, Taverna C.
Eosinophilic pneumonia after administration of fludarabine for the
treatment of non-Hodgkin’s lymphoma. Ann
Hematol. 2002;81:535-537.
-
Yamakado S, Yoshida Y, Yamada T, Kishida T, Kobayashi M, Nomura T.
Pulmonary infiltration and eosinophilia associated with sulfasalazine
therapy for ulcerative colitis: a case report and review of
literature. Intern Med. 1992;31:108-113.
-
Clauw DJ, Nashel DJ, Umhau A, Katz P.
Tryptophan-associated eosinophilic connective-tissue disease: a new
clinical entity? JAMA. 1990;263:1502-1506.
-
Kilbourne EM, Posada
de la Paz M, Abaitua
Borda I, Diez
Ruiz-Navarro M, Philen RM, Falk H.
Toxic oil syndrome: a current clinical and epidemiologic summary, including
comparisons with the eosinophilia-myalgia syndrome. J Am Coll
Cardiol. 1991;18:711-717.
-
Mert A, Tabak F, Aktuglu Y.
Eosinophilia in toxic shock syndrome: review of 20 cases
[letter]. Scand J Infect Dis. 1998;30:320.
-
Griffin E, Hakansson L, Formgren H, Jorgensen K, Peterson C, Venge P.
Blood eosinophil number and activity in relation to lung function in
patients with asthma and with eosinophilia. J Allergy Clin
Immunol. 1991;87:548-557.
-
Uehara M, Izukura R, Sawai T.
Blood eosinophilia in atopic dermatitis. Clin Exp
Dermatol. 1990;15:264-266.
-
Chapman BJ, Capewell S, Gibson R, Greening AP, Crompton GK.
Pulmonary eosinophilia with and without allergic bronchopulmonary
aspergillosis. Thorax. 1989;44:919-924.
-
Desenne JJ, Acquatella G, Stern R, Muller A, Sanchez M, Somoza R.
Blood eosinophilia in Hodgkin’s disease: a follow-up of 25 cases
in Venezuela. Cancer. 1992;69:1248-1253.
-
Scales JW, McMichael A.
Persistent peripheral eosinophilia and cutaneous non-Hodgkin’s
lymphoma: a case report and review of the
literature. Cutis. 2001;67:67-70.
-
Watanabe K, Shinbo T, Kojima M, Naito M, Tanahashi N, Nara M.
Bcell lymphoma associated with
eosinophilia. Cancer. 1989;64:1682-1685.
-
Navarro-Roman L, Medeiros LJ, Kingma DW, et
al. Malignant lymphomas of B-cell lineage with marked tissue eosinophilia:
a report of five cases. Am J Surg
Pathol. 1994;18:347-356.
-
Fridlender ZG, Simon HU, Shalit M.
Metastatic carcinoma presenting with concomitant eosinophilia and
thromboembolism. Am J Med Sci. 2003;326:98-101.
-
Walter R, Joller-Jemelka HI, Salomon F.
Metastatic squamous cell carcinoma with marked blood eosinophilia and
elevated serum interleukin-5 levels [letter]. Exp
Hematol. 2002;30:1-2.
-
Spry C. Eosinophilia in Addison’s
disease. Yale J Biol Med. 1976;49:411-413.
-
Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD.
Human eotaxin is a specific chemoattractant for eosinophil cells and
provides a new mechanism to explain tissue eosinophilia. Nat
Med. 1996;2:449-456.
-
Teruya-Feldstein J, Jaffe ES, Burd PR, Kingma DW, Setsuda JE, Tosato G.
Differential chemokine expression in tissues involved by Hodgkin’s
disease: direct correlation of eotaxin expression and tissue
eosinophilia. Blood. 1999;93:2463-2470.
-
Balducci L, Chapman SW, Little DD, Hardy CL.
Paraneoplastic eosinophilia: report of a case with in vitro studies
of hemopoiesis. Cancer. 1989;64:2250-2253.
-
Sanada I, Asou N, Kojima S, Kawano F, Shido T, Takatsuki K.
Acute myelogenous leukemia (FAB M1) associated with t(5;16) and
eosinophilia: report of an additional case. Cancer Genet
Cytogenet. 1989;43:139-141.
-
Blatt PM, Rothstein G, Miller HL, Cathey WJ.
Loffler’s endomyocardial fibrosis with eosinophilia in association
with acute lymphoblastic
leukemia. Blood. 1974;44:489-493.
-
Keung YK, Beaty M, Steward W, Jackle B, Pettnati M.
Chronic myelocytic leukemia with eosinophilia, t(9;12)(q34;p13), and
ETV6-ABL gene rearrangement: case report and review of the
literature. Cancer Genet Cytogenet. 2002;138:139-142.
-
Kuroda J, Kimura S, Akaogi T, et
al. Myelodysplastic syndrome with clonal eosinophilia accompanied by
eosinophilic pulmonary interstitial infiltration. Acta
Haematol. 2000;104:119-123.
-
Wilkinson K, Velloso ER, Lopes LF, et
al. Cloning of the t(1;5)(q23;q33) in a myeloproliferative disorder
associated with eosinophilia: involvement of PDGFRB and response to
imatinib. Blood. 2003;102:4187-4190.
-
Bain BJ. Cytogenetic and molecular
genetic aspects of eosinophilic leukaemias. Br J
Haematol. 2003;122:173-179.
-
Ma SK, Kwong YL, Shek TW, et
al. The role of trisomy 8 in the pathogenesis of chronic eosinophilic
leukemia. Hum Pathol. 1999;30:864-868.
-
Yam LT, Yam CF, Li CY.
Eosinophilia in systemic mastocytosis. Am J Clin
Pathol. 1980;73:48-54.
-
Bennett JM, Catovsky D, Daniel MT, et
al. The chronic myeloid leukaemias: guidelines for distinguishing chronic
granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia:
proposals by the French-American-British Cooperative Leukaemia
Group. Br J Haematol. 1994;87:746-754.
-
Heldin CH, Westermark B.
Mechanism of action and in vivo role of platelet-derived growth
factor. Physiol Rev. 1999;79:1283-1316.
-
Pardanani A, Brockman SR, Paternoster SF, et
al. FIP1L1-PDGFRA fusion: prevalence and clinicopathologic correlates
in 89 consecutive patients with moderate to severe
eosinophilia. Blood. 2004;104:3038-3045.
-
Elliott MA, Pardanani A, Li CY, Tefferi A.
Immunophenotypic normalization of aberrant mast cells accompanies
histological remission in imatinibtreated patients with eosinophilia-associated
mastocytosis [letter]. Leukemia. 2004;18:1027-1029.
-
Pardanani A, Ketterling RP, Brockman SR, et
al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in
systemic mastocytosis associated with eosinophilia and predicts response
to imatinib mesylate
therapy. Blood. 2003;102:3093-3096.
-
Cools J, DeAngelo DJ, Gotlib J, et
al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes
as a therapeutic target of imatinib in idiopathic hypereosinophilic
syndrome. N Engl J Med. 2003;348:1201-1214.
-
Baxter EJ, Hochhaus A, Bolufer P, et
al. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses
BCR to PDGFRA. Hum Mol Genet. 2002;11:1391-1397.
-
Miranda RN, Esparza AR, Sambandam S, Medeiros LJ.
Systemic mast cell disease presenting with peripheral blood
eosinophilia. Hum Pathol. 1994;25:727-730.
-
Klion AD, Robyn J, Akin C, et
al. Molecular remission and reversal of myelofibrosis in response to
imatinib mesylate treatment in patients with the myeloproliferative variant
of hypereosinophilic
syndrome. Blood. 2004;103:473-478.
-
Vandenberghe P, Wlodarska I, Michaux L, et
al. Clinical and molecular features of FIP1L1-PDFGRA (+) chronic
eosinophilic leukemias. Leukemia. 2004;18:734-742.
-
Golub TR, Barker GF, Lovett M, Gilliland DG.
Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic
myelomonocytic leukemia with t(5;12) chromosomal
translocation. Cell. 1994;77:307-316.
-
Granjo E, Lima M, Lopes JM, et
al. Chronic eosinophilic leukaemia presenting with erythroderma, mild
eosinophilia and hyper-IgE: clinical, immunological and cytogenetic features
and therapeutic approach: a case report. Acta
Haematol. 2002;107:108-112.
-
Apperley JF, Gardembas M, Melo JV, et
al. Response to imatinib mesylate in patients with chronic
myeloproliferative diseases with rearrangements of the platelet-derived growth
factor receptor beta. N Engl J Med. 2002;347:481-487.
-
Gupta R, Knight CL, Bain BJ.
Receptor tyrosine kinase mutations in myeloid neoplasms. Br
J Haematol. 2002;117:489-508.
-
Macdonald D, Reiter A, Cross NC.
The 8p11 myeloproliferative syndrome: a distinct clinical entity caused
by constitutive activation of FGFR1. Acta
Haematol. 2002;107:101-107.
-
Sohal J, Chase A, Mould S, et
al. Identification of four new translocations involving FGFR1 in myeloid
disorders. Genes Chromosomes Cancer. 2001;32:155-163.
-
Fauci AS, Harley JB, Roberts WC, Ferrans VJ, Gralnick HR, Bjornson BH.
The idiopathic hypereosinophilic syndrome: clinical, pathophysiologic,
and therapeutic considerations [NIH conference]. Ann Intern
Med. 1982;97:78-92.
-
Liesveld JL, Abboud CN.
State of the art: the hypereosinophilic syndromes. Blood
Rev. 1991;5:29-37.
-
Lin DA, Boyce JA.
The idiopathic hypereosinophilic syndrome. Allergy Asthma
Proc. 2003;24:417-420.
-
Owen J, Scott JG.
Transition of the hypereosinophilic syndrome to myelomonocytic
leukemia. Can Med Assoc J. 1979;121:1489-1491.
-
Yoo TJ, Orman SV, Patil SR, et
al. Evolution to eosinophilic leukemia with a t(5:11) translocation
in a patient with idiopathic hypereosinophilic syndrome. Cancer Genet
Cytogenet. 1984;11:389-394.
-
Needleman SW, Mane SM, Gutheil JC, Kapil V, Heyman MR, Testa JR.
Hypereosinophilic syndrome with evolution to myeloproliferative disorder:
temporal relationship to loss of Y chromosome and c-N-ras
activation. Hematol Pathol. 1990;4:149-155.
-
Chang HW, Leong KH, Koh DR, Lee SH.
Clonality of isolated eosinophils in the hypereosinophilic
syndrome. Blood. 1999;93:1651-1657.
-
Roufosse F, Schandene L, Sibille C, et
al. Clonal Th2 lymphocytes in patients with the idiopathic hypereosinophilic
syndrome. Br J Haematol. 2000;109:540-548.
-
Raghavachar A, Fleischer S, Frickhofen N, Heimpel H, Fleischer B.
T lymphocyte control of human eosinophilic granulopoiesis: clonal analysis
in an idiopathic hypereosinophilic syndrome. J
Immunol. 1987;139:3753-3758.
-
Simon HU, Plotz SG, Dummer R, Blaser K.
Abnormal clones of T cells producing interleukin-5 in idiopathic
eosinophilia. N Engl J Med. 1999;341:1112-1120.
-
Spry CJ, Davies J, Tai PC, Olsen EG, Oakley CM, Goodwin JF.
Clinical features of fifteen patients with the hypereosinophilic
syndrome. Q J Med. 1983;52:1-22.
-
Kazmierowski JA, Chusid MJ, Parrillo JE, Fauci AS, Wolff SM.
Dermatologic manifestations of the hypereosinophilic
syndrome. Arch Dermatol. 1978;114:531-535.
-
Leiferman KM, O’Duffy JD, Perry HO, Greipp PR, Giuliani ER, Gleich GJ.
Recurrent incapacitating mucosal ulcerations: a prodrome of the
hypereosinophilic
syndrome. JAMA. 1982;247:1018-1020.
-
Bogenrieder T, Griese DP, Schiffner R, et
al. Wells’ syndrome associated with idiopathic hypereosinophilic
syndrome. Br J Dermatol. 1997;137:978-982.
-
Parrillo JE, Borer JS, Henry WL, Wolff SM, Fauci AS.
The cardiovascular manifestations of the hypereosinophilic syndrome:
prospective study of 26 patients, with review of the literature. Am
J Med. 1979;67:572-582.
-
Sato Y, Taniguchi R, Yamada T, et
al. Measurement of serum concentrations of cardiac troponin T in patients
with hypereosinophilic syndrome: a sensitive non-invasive marker of cardiac
disorder [letter]. Intern Med. 2000;39:350.
-
Dorfman LJ, Ransom BR, Forno LS, Kelts A.
Neuropathy in the hypereosinophilic syndrome. Muscle
Nerve. 1983;6:291-298.
-
Moore PM, Harley JB, Fauci AS.
Neurologic dysfunction in the idiopathic hypereosinophilic
syndrome. Ann Intern Med. 1985;102:109-114.
-
Weingarten JS, O’Sheal SF, Margolis WS.
Eosinophilic meningitis and the hypereosinophilic syndrome: case report
and review of the literature. Am J
Med. 1985;78:674-676.
-
Tsai CP, Yeh HH, Tsai JJ, Lin KP, Wu ZA.
Transverse myelitis and polyneuropathy in idiopathic hypereosinophilic
syndrome [letter]. Muscle Nerve. 1993;16:112-113.
-
Slabbynck H, Impens N, Naegels S, Dewaele M, Schandevyl W.
Idiopathic hypereosinophilic syndrome-related pulmonary involvement
diagnosed by bronchoalveolar
lavage. Chest. 1992;101:1178-1180.
-
Winn RE, Kollef MH, Meyer JI.
Pulmonary involvement in the hypereosinophilic
syndrome. Chest. 1994;105:656-660.
-
Kang EY, Shim JJ, Kim JS, Kim KI.
Pulmonary involvement of idiopathic hypereosinophilic syndrome: CT
findings in five patients. J Comput Assist
Tomogr. 1997;21:612-615.
-
Scheurlen M, Mork H, Weber P.
Hypereosinophilic syndrome resembling chronic inflammatory bowel disease
with primary sclerosing cholangitis. J Clin
Gastroenterol. 1992;14:59-63.
-
Catalano L, Pastena S, Catalano M, Lestini A, Valentini G, Rotoli B.
Eosinophilic gastroenteritis: a rare localization of the hypereosinophilic
syndrome? [letter]. Haematologica. 1992;77:292-293.
-
Grauer L, Padilla VMIII, Bouza L, Barkin JS.
Eosinophilic sclerosing cholangitis associated with hypereosinophilic
syndrome. Am J Gastroenterol. 1993;88:1764-1769.
-
Ichikawa N, Taniguchi A, Akama H, et
al. Sclerosing cholangitis associated with hypereosinophilic
syndrome. Intern Med. 1997;36:561-564.
-
Elouaer-Blanc L, Zafrani ES, Farcet JP, Saint-Marc Girardin
MF, Mathieu D, Dhumeaux D.
Hepatic vein obstruction in idiopathic hypereosinophilic
syndrome. Arch Intern Med. 1985;145:751-753.
-
Vargas CA, Maldonado O, Botero RC, et
al. Budd-Chiari syndrome associated with the hypereosinophilic syndrome
[letter]. Am J Gastroenterol. 1993;88:1802-1803.
-
Chaine G, Davies J, Kohner EM, Hawarth S, Spry CJ.
Ophthalmologic abnormalities in the hypereosinophilic
syndrome. Ophthalmology. 1982;89:1348-1356.
-
Peison B, Benisch B, Lim M, Chin JC.
Idiopathic hypereosinophilic syndrome with polymyositis. South
Med J. 1988;81:403-406.
-
Bulucu F, Can C, Inal V, Baykal Y, Erikci S.
Renal involvement in a patient with idiopathic hypereosinophilic syndrome
[letter]. Clin Nephrol. 2002;57:171-173.
-
Prattichizzo FA, Bernini L.
An idiopathic hypereosinophilic syndrome mimicking seronegative rheumatoid
arthritis: 20-year follow-up with clinical and laboratory
findings. Clin Exp Rheumatol. 1992;10:79-81.
-
Appelbaum FR. Molecular diagnosis
and clinical decisions in adult acute leukemia. Semin
Hematol. 1999;36:401-410.
-
O’Brien SG, Guilhot F, Larson RA, et
al IRIS Investigators. Imatinib compared with interferon and low-dose
cytarabine for newly diagnosed chronic-phase chronic myeloid
leukemia. N Engl J Med. 2003;348:994-1004.
-
Druker BJ, Tamura S, Buchdunger E, et
al. Effects of a selective inhibitor of the Abl tyrosine kinase on the
growth of Bcr-Abl positive cells. Nat
Med. 1996;2:561-566.
-
Buchdunger E, Zimmermann J, Mett H, et
al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo
by a 2-phenylaminopyrimidine derivative. Cancer
Res. 1996;56:100-104.
-
Deininger MW, Goldman JM, Lydon N, Melo JV.
The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth
of BCR-ABL-positive cells. Blood. 1997;90:3691-3698.
-
Carroll M, Ohno-Jones S, Tamura S, et
al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells
expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion
proteins. Blood. 1997;90:4947-4952.
-
Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ.
Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a
selective tyrosine kinase
inhibitor. Blood. 2000;96:925-932.
-
Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J.
Structural mechanism for STI-571 inhibition of abelson tyrosine
kinase. Science. 2000;289:1938-1942.
-
Tefferi A, Pardanani A.
Clinical, genetic, and therapeutic insights into systemic mast cell
disease. Curr Opin Hematol. 2004;11:58-64.
-
Takeuchi K, Koike K, Kamijo T, et
al. STI571 inhibits growth and adhesion of human mast cells in
culture. J Leukoc Biol. 2003;74:1026-1034.
-
Akin C, Brockow K, D’Ambrosio C, et
al. Effects of tyrosine kinase inhibitor STI571 on human mast cells
bearing wild-type or mutated c-kit. Exp
Hematol. 2003;31:686-692.
-
Zermati Y, De
Sepulveda P, Feger F, et
al. Effect of tyrosine kinase inhibitor STI571 on the kinase activity
of wild-type and various mutated c-kit receptors found in mast cell
neoplasms. Oncogene. 2003;22:660-664.
-
Pardanani A, Elliott M, Reeder T, et
al. Imatinib for systemic mast-cell
disease. Lancet. 2003;362:535-536.
-
Dispenzieri A, Kyle RA, Lacy MQ, et
al. Superior survival in primary systemic amyloidosis patients undergoing
peripheral blood stem cell transplantation: a case-control
study. Blood. 2004;103:3960-3963.
-
Martinelli G, Malagola M, Ottaviani E, Rosti G, Trabacchi E, Baccarani M.
Imatinib mesylate can induce complete molecular remission in FIP1L1-PDGFR-a
positive idiopathic hypereosinophilic syndrome
[letter]. Haematologica. 2004;89:236-237.
-
Frickhofen N, Marker-Hermann E, Reiter A, et
al. Complete molecular remission of chronic eosinophilic leukemia
complicated by CNS disease after targeted therapy with imatinib. Ann
Hematol. 2004;83:477-480.
-
Rose C, Dupire S, Roche-Lestienne C, et
al. Sustained molecular response with imatinib in a leukemic form of
idiopathic hypereosinophilic syndrome in relapse after allograft
[letter]. Leukemia. 2004;18:354-355.
-
Pitini V, Arrigo C, Teti D, Barresi G, Righi M, Alo G.
Response to STI571 in chronic myelomonocytic leukemia with platelet
derived growth factor beta receptor involvement: a new case
report. Haematologica. 2003;88:ECR18.
-
Parrillo JE, Fauci AS, Wolff SM.
Therapy of the hypereosinophilic syndrome. Ann Intern
Med. 1978;89:167-172.
-
Schooley RT, Flaum MA, Gralnick HR, Fauci AS.
A clinicopathologic correlation of the idiopathic hypereosinophilic
syndrome, II: clinical
manifestations. Blood. 1981;58:1021-1026.
-
Zabel P, Schlaak M.
Cyclosporin for hypereosinophilic syndrome. Ann
Hematol. 1991;62:230-231.
-
Sakamoto K, Erdreich-Epstein A, deClerck Y, Coates T.
Prolonged clinical response to vincristine treatment in two patients
with idiopathic hypereosinophilic syndrome. Am J Pediatr Hematol
Oncol. 1992;14:348-351.
-
Marshall GM, White L.
Effective therapy for a severe case of the idiopathic hypereosinophilic
syndrome. Am J Pediatr Hematol Oncol. 1989;11:178-183.
-
Fruehauf S, Fiehn C, Haas R, Doehner H, Hunstein W.
Sustained remission of idiopathic hypereosinophilic syndrome following
alpha-interferon therapy. Acta Haematol. 1993;89:91-93.
-
Butterfield JH, Gleich GJ.
Response of six patients with idiopathic hypereosinophilic syndrome
to interferon alfa. J Allergy Clin Immunol. 1994;94(6, pt
2):1318-1326.
-
Ceretelli S, Capochiani E, Petrini M.
Interferon-a in the idiopathic hypereosinophilic syndrome: consideration
of five cases. Ann Hematol. 1998;77:161-164.
-
Ueno NT, Zhao S, Robertson LE, Consoli U, Andreeff M.
2-Chlorodeoxyadenosine therapy for idiopathic hypereosinophilic syndrome
[letter]. Leukemia. 1997;11:1386-1390.
-
Smit AJ, van
Essen LH, de Vries EG.
Successful long-term control of idiopathic hypereosinophilic syndrome
with etoposide. Cancer. 1991;67:2826-2827.
-
Sigmund DA, Flessa HC.
Hypereosinophilic syndrome: successful allogeneic bone marrow
transplantation. Bone Marrow
Transplant. 1995;15:647-648.
-
Fukushima T, Kuriyama K, Ito H, et
al. Successful bone marrow transplantation for idiopathic hypereosinophilic
syndrome. Br J Haematol. 1995;90:213-215.
-
Juvonen E, Volin L, Koponen A, Ruutu T.
Allogeneic blood stem cell transplantation following non-myeloablative
conditioning for hypereosinophilic syndrome. Bone Marrow
Transplant. 2002;29:457-458.
-
Ueno NT, Anagnostopoulos A, Rondon G, et
al. Successful non-myeloablative allogeneic transplantation for treatment
of idiopathic hypereosinophilic syndrome. Br J
Haematol. 2002;119:131-134.
-
Klion AD, Law MA, Noel P, Kim YJ, Haverty TP, Nutman TB.
Safety and efficacy of the monoclonal anti-interleukin-5 antibody SCH55700
in the treatment of patients with hypereosinophilic
syndrome. Blood. 2004;103:2939-2941.
-
Garrett JK, Jameson SC, Thomson B, et
al. Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic
syndromes. J Allergy Clin Immunol. 2004;113:115-119.
-
Plotz SG, Simon HU, Darsow U, et
al. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome
with eosinophilic dermatitis. N Engl J
Med. 2003;349:2334-2339.
-
Sefcick A, Sowter D, DasGupta E, Russell NH, Byrne JL.
Alemtuzumab therapy for refractory idiopathic hypereosinophilic syndrome
[letter]. Br J Haematol. 2004;124:558-559.
-
Koury MJ, Newman JH, Murray JJ.
Reversal of hypereosinophilic syndrome and lymphomatoid papulosis with
mepolizumab and imatinib [letter]. Am J
Med. 2003;115:587-589.
-
Pardanani A, Reeder T, Porrata LF, et
al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic
disorders. Blood. 2003;101:3391-3397.
-
Pitini V, Arrigo C, Azzarello D, et
al. Serum concentration of cardiac Troponin T in patients with
hypereosinophilic syndrome treated with imatinib is predictive of adverse
outcomes [letter]. Blood. 2003;102:3456-3457.
-
Gleich GJ, Leiferman KM, Pardanani A, Tefferi A, Butterfield JH.
Treatment of hypereosinophilic syndrome with imatinib
mesilate. Lancet. 2002;359:1577-1578.
-
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL.
Overriding imatinib resistance with a novel ABL kinase
inhibitor. Science. 2004;305:399-401.
-
Tipping AJ, Baluch S, Barnes DJ, et
al. Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors
in cells sensitive and resistant to imatinib
mesylate. Leukemia. 2004;18:1352-1356.
-
-
Back to top |
Home Page
{kind=link}